Intrinsically Disordered Proteins (IDPs): Conformational Dynamics and Interactions
Unlike the funnel-shaped landscape of globular proteins, IDPs have a rather shallow but rugged energy landscape, with many local minima separated by small barriers. They interact, often tightly and specifically, with several partners in so-called "hubs", but the physics of this process (kinetics, energetics, molecular forces) is underdetermined. Around 65% of the signaling and 75% of the cancer-associated proteins are predicted to have disordered regions, thus implying an important role for IDPs in mediating regulatory interactions in biological processes.
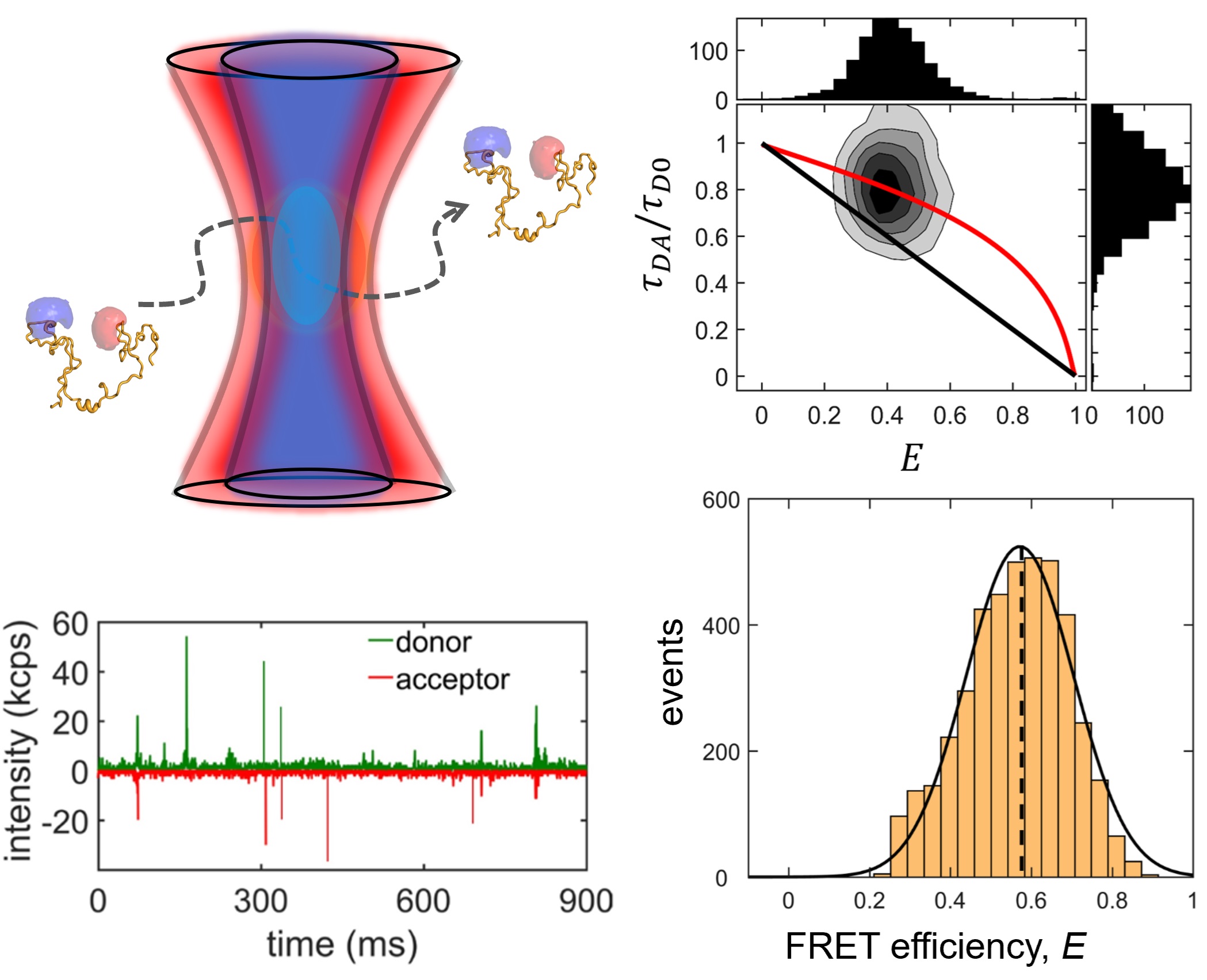
We study two IDP systems: Sic1, a kinase inhibitor in the yeast the cell cycle, and 4E-BP2, a translation inhibitor involved in regulating synaptic plasticity. Sic1 forms a dynamic "fuzzy" complex with the WD40 domain of the Cdc4 protein, where the binding affinity shows an intriguing non-linear dependence on the number of phosphate groups on Sic1. 4E-BP2 acts like a regulatory switch: in the non-phosphorylated state is disordered and bound to the initiation factor eIF4E, whereas in the phosphorylated state it partially folds and detaches, allowing another protein to dock on eIF4E and initiate translation. We use single-molecule fluorescence spectroscopy to study these two IDP complexes, to understand how "binding without folding" occurs in biology.
G Protein Coupled Receptors (GPCRs): Implications of Dynamics and Oligomerization for Cellular Signalling
GPCRs constitute the largest superfamily of proteins encoded by mammalian genomes. They serve as transducers of the signal between extracellular ligands such as hormones or drugs and intracellular mediators such as G proteins and arrestins. This makes GPCRs to the targets of more than 30% of all modern drugs, with the potential to treat many diseases, such as diabetes, Parkinson's, cardiovascular disease, depression, drug addiction and obesity.
: Ligands, Allostery, Signaling and Oligomers")
Our lab is studying two GPCRs: muscarinic cholinergic receptors, involved in cognitive functions in the brain (M1) or modulate heart rate and muscle contraction (M2), and the A2A adenosine receptor, which regulates dopamine release in the brain. Our single-molecule techniques (photobleaching counting, single-particle tracking and dual-color FCS) can determine the presence and size of receptor oligmers, and delineate their role for cellular signalling. Using smFRET, we can characterize the effect of ligands on the conformational states and dynamics of GPCRs, with the aim to resolve different activation pathways and reveal the mechanism of biased signalling. The coupling between receptors and G proteins can visualized at single-molecule level in real time in either controlled in vitro preparations or in the native environment of the live cell. We corroborate our single-molecule data with magnetic resonance and molecular dynamics from our collaborators to gain new insights into allostery, cooperativity and signalling bias of GPCRs.
Ensemble Descriptions of Disordered Proteins: Integrating Experiments and Computations
The potentially vast number of relevant structures of IDPs makes their biophysical characterization difficult. Modelling them necessitates a framework of sufficient complexity to capture important features, while avoiding being too large to be computationally intractable. IDPs are often modelled as conformational ensembles, which are a set of atomistic 3D structures with associated statistical weights. Our group used data from nuclear magnetic resonance (NMR), small-angle X-ray scattering (SAXS), and single-molecule Förster resonance energy transfer (smFRET) to refine a starting pool of conformations. Different experiments are sensitive to different length scales and timescales, with different degrees of time-averaging and ensemble-averaging.

Increasingly reliable conformational ensembles have been obtained for isolated IDPs, by using more sophisticated conformer generators and more robust optimization algorithms. The next step is to calculate ensembles for disordered proteins in complex with their biological targets and explore the underlying low-dimensional space describing their conformational fluctuations. Clustering and dimensionality reduction techniques applied to optimized Sic1:Cdc4 and BP2:4E ensembles will quantify the average structural statistics of prevalent states of these two highly dynamic IDP complexes. This integrative computational work will formulate new hypotheses to define IDP interactions and inform the design of drugs against their pathological activity.